tangxie520 第29页
-

fda登记_FDA注册登记证书
FDA登记系统:FDA登记系统 (FIS) 的创建旨在促进向美国食品和药物管理局 (FDA) 的提交,包括注册、列表和其他通知。自美国东部时间2003年10月16日下午6:00起,FIS 每周 7 天、每天24小时可用。FIS的创建部分是为了响应2002年的《生物恐怖主义法案》,该法案高度重视改进信息管理以帮助保护食品供应。该法案要求 FDA 开发了两个系统:一个支持在美国生产、加工、包装或保存拟在美国消费的食品的设施的注册,另一个在食品进口或提供进口到美国之前收到事先通知。根据美国法律,相关产品出口必须完成FDA...
-

什么是药品FDA注册代码 (NDC),它是如何分配的,要求是什么?
什么是药品NDC注册?为了跟踪,药品被分配了一个唯一的标识符,称为国家药品代码或 NDC 编号。制药公司必须注册其生产设施并列出商业分销的药物。谁分配NDC号?美国食品和药物管理局(FDA),NDC 编号是分配给药物的10位数字代码,NDC 的前五位数字来自贴标代码。FDA将标签代码分配给产品的制造商、分销商或私人标签分销商。NDC 编号的第一步是从FDA获得标签代码。NDC编号格式:NDC 编号可以以 5 -4-1 或 5 -3-2 格式分配,这意味着您可以分配 4 位或 3 位代码产品和 1 位或 2 位包装代...
-

什么是FDA表格2877?激光辐射产品FDA注册后出口要求
什么是FDA表格2877? 辐射电子产品受FDA监管,并且必须符合21CFR1000-1005中的一般要求,先完成FDA激光注册。食品和药物管理局(FDA)2877表格是进口电子产品(及其零件)受FDA辐射控制标准约束的声明。进口商负责签署并提供证明进口货物符合FDA要求的表格。需要符合FDA2877的产品示例包括但不限于:微波炉、阴极射线管、激光打印机和CD播放器。我什么时候必须提交FDA-2877表格? 符合美国联邦性能标准的辐射电子...
-

食品FDA预先通知是啥?什么时候必须向FDA提交事先通知?
什么是事先通知FDA? 食品到达美国之前向美国食品和药物管理局 (FDA) 通报进口食品,包括有关产品、数量和包装以及相关设施的信息,例如制造商、托运人、所有者和最终收货人。每一批货都需要做预先通知(一般是食品才要),物流发出后提交(需要运单号),先做FDA注册,要不然清不了关。商通检测提供FDA注册和预先通知(提前海关报备)服务!FDA进口食品事前通知介绍: 2002 年《公共卫生安全和生物恐怖主义防范和应对法》(《生物恐怖主义法》)...
-
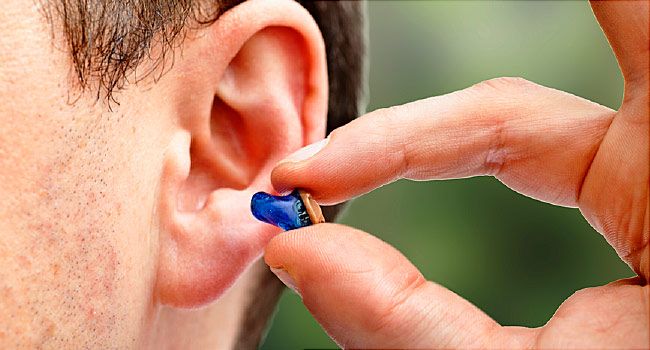
助听器FAD认证_哪些助听器才能做FDA医疗注册?
超过3500万美国人患有某种形式的听力损失,助听器出口美国是一个巨大的市场,助听器出口美国是属于医疗器械,需要强制FDA认证才能顺利进入美国市场,商通检测提供助听器FDA认证医疗注册服务!助听器FDA认证是什么意思? 美国食品和药物管理局 (FDA) 是一个正府机构,负责监管国内外制造的食品和饮料、药物和医疗器械的生产,并根据可靠性、安全性和性能标准对其进行测试。它还规范助听器,旨在为单耳或双耳轻度至重度听力损失的个人提供助听器的医疗设备。助听器与非处方 (OTC) 声音放大器...
-

FDA注册续费_食品、医疗、药品FDA认证有效期到了怎么更新
FDA注册续费介绍: 美国食品和药物管理局(FDA)要求生产和分销在美国境内使用的产品的“医疗器械”和“药品企业”每年更新其FDA注册,这些企业必须在每年年10月1日至12月31日之间续签更新他的FDA注册,过了时间FDA会从数据库中删除其注册信息,商通检测可以协助更新您的医疗器械或药品企业FDA注册!关于FDA注册的有效期:1.普通食品FDA:偶数年年底更新注册,最长有效期2年,2.酸化食品FDA,企业注册FCE偶数年底更新注册,产品注册SID一直有效3.医疗器械FDA,当...
-
FDA认证_医疗器械再加工指南:验证和FDA审查
美国食品和药物管理局(FDA 或该机构)是美国医疗器械领域的监管机构,已发布了一份指导文件,专门针对医疗机构中的医疗器械进行再处理。该指南旨在帮助参与医疗器械操作的各方确保遵守与为同一患者或其他患者在使用后和使用前对医疗器械进行再处理相关的监管要求。本文重点审查监管机构提交的再处理说明和再处理方法验证文件。由于其法律性质,该文件没有引入新的规则和要求,但对现有立法条款的解释方式提供了额外的澄清,以及需要考虑的额外建议。这些要求不具有约束力,因此,可以应用替代方法,前提是这种方法符合适用的监管要求并事先与当局达成一致...
-

如何为FDA认证检查准备设计历史文件
食品和药物管理局 (FDA认证) 的检查压力很大,尤其是当您的文件未准备好进行审计时。创建和维护合规设计历史文件(DHF) 将有助于确保当时机成熟时,您将准备好接受FDA检查,从而将结果降至最低。缺乏DHF的情况并不少见,尤其是当产品开发过程没有特别强调监管要求时。设备设计人员专注于他们最擅长的事情,不一定在此过程中优先考虑记录设计控制。你越主动,出现的问题就越少。不要等到突击检查才知道您的DHF需要一些工作。包含在您的设计历史文件中的元素DHF 是设备设计和开发的完整记录——“完整”是这里的关键词。FDA 正在寻...
-

FDA认证医疗器械510(k)豁免查询和GMP要求
介绍:以下是按设备类别列出的510(k)豁免和良好生产规范(GMP)/质量体系豁免的细分。除非通过脚注进一步限定,否则此列表中的所有设备均免除510(k)。除了一般记录保存要求和合规文件外,只有带(*)注释的设备也免于GMP。I类设备FDA认证: FDA已经豁免了几乎所有I类器械(保留器械除外,其上市前通知要求除外,包括那些被1994年12月7日和1996年1月16日联邦公报中公布的最终法规豁免的器械。一些510(k)用“\#\”注释的豁免具有脚注中指出的某些限制。重要的是确认豁免状态和适...
-
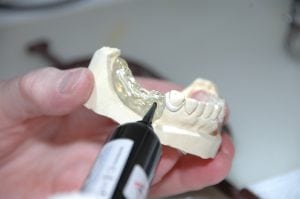
牙科产品FDA认证哪些是医疗510k注册怎么分类?
FDA是否监管牙科设备? 对于牙医和牙科实验室,美国食品和药物管理局(FDA)以某种方式对用于提供牙科服务的原材料和设备/技术进行了监管。为了使医疗器械在美国合法销售(销售),它们必须得到FDA的批准或批准,除非它是510(k)豁免的。部分牙科设备是按照一类医疗注册FDA,有些需要按照2类医疗510K注册,评估产品具体分类可联系商通检测!已批准的医疗器械:这些医疗器械是FDA已确定与另一合法销售的器械实质上等效(类似)的器械。上市前通知提交被称为510(k),必须提交给FDA...
-

FDA认证发布首个OTC专论最终订单;包括防晒药品要求更新
2021年9月20日,美国食品药品监督管理局(FDA认证)发布了首批针对非处方药(OTC)药品专论的最终行政命令,这是OTC专论改革中最先出现的。FDA于2020年3月7日根据《冠状病毒援助、救济和经济安全(CARES)法案》制定了改革措施。FDA发布了以下第一批最终行政命令:1.非处方人用夜间睡眠辅助药品2.非处方人用抗气胀药物产品3.供非处方人使用的局部耳部药物产品4.非处方人用玉米和愈伤组织去除剂药品自第一批以来,FDA继续发布更多最终行政命令,最近添加的产品包括用于非处方人类使用的防晒药物产品的最终订单。防...
-

FDA认证支持从色素添加剂列表中去除醋酸铅
美国食品和药物管理局(FDA认证)将继续从用于头皮染发的化妆品中的着色添加剂认证订单中废除醋酸铅。对题为“终止豁免认证的色素添加剂订单”的最终规则的保留;醋酸铅”是为了回应染发剂制造商CombeInc.对该规则的反对。在评估CombeInc.的反对意见并发现没有理由推翻裁决后,FDA将支持对颜色添加剂订单的修订,不再规定在染发剂中使用醋酸铅。FDA认证颜色添加剂的最终规则:FDA的最终规则于2018年10月31日发布修改了颜色添加剂法规,以反映FDA的决定,即“不再有合理的确定性认为,在用于染发和头皮的化妆品中添加...
-
医疗和药品FDA注册必须在21年10月1日至12月31日间完成更新
医疗器械和药品公司必须在2021年10月1日至12月31日之间更新续费其FDA注册美国食品和药物管理局(FDA认证)要求生产和分销在美国境内使用的产品的医疗器械和药品企业每年更新其FDA注册。这些企业必须在2021年10月1日至2021年12月31日之间续签2022年的注册。医疗器械机构FDA注册更新要求: FDA注册的医疗器械企业必须指定一名负责年度注册的官方通讯员。位于美国境外的医疗器械机构还必须指定美国代理商。医疗器械企业必须在FDA统一注册和列表系统(FURLS)中列出...
-

激光笔FDA认证注册发射功率有强制性限制
本文解释了适用于激光笔的FDA认证监管要求,解释了这些产品的功率限制,以及安全使用信息和宣传材料的适用要求。还讨论了对高功率、电池供电的便携式激光器的不当使用。激光产品FDA认证是如何分类的?激光器通常根据其发出的光的数量和类型所造成的危害进行分类。危险等级范围从I类到IV类,其中I类激光是无害的,而IV类激光是最危险的。I类产品包括激光打印机和CD播放器,其中激光辐射通常包含在产品中。超过I类的产品允许接触一定量的激光辐射。II类和IIa类产品包括条形码扫描仪。IIIa类产品包括激光笔。IIIb和IV类产品包括激...
-

指甲油美甲产品美国FDA注册监管要求
指甲油获得FDA批准?指甲油属于化妆品,指甲油和指甲油去除剂必须是安全的——并且受到美国食品和药物管理局的监管。出口美国不强制FDA认证,但是亚马逊是强制要求FDA注册,FDA还监管用于干燥(或“治愈”)人造指甲或凝胶指甲油作为电子产品的设备,因为它们会发出辐射。家庭和美发店使用的美甲产品由食品和药物管理局监管,根据联邦食品、药品和化妆品法案(FD&C 法案),这些产品通常作为化妆品进行监管 [FD&C 法案,第 201(i) 节]。用于治疗医疗问题(例如指甲真菌)的美甲产品是药物。商通检测提供化妆...
-

 关于商通检测
关于商通检测商通检测是一家第三方认证咨询公司,为国内外企业提供全方位法规咨询服务。如果您正在寻求产品认证法规帮助,商通检测是您不错的选择,我们经验丰富的工程师可以为您提供指导。